
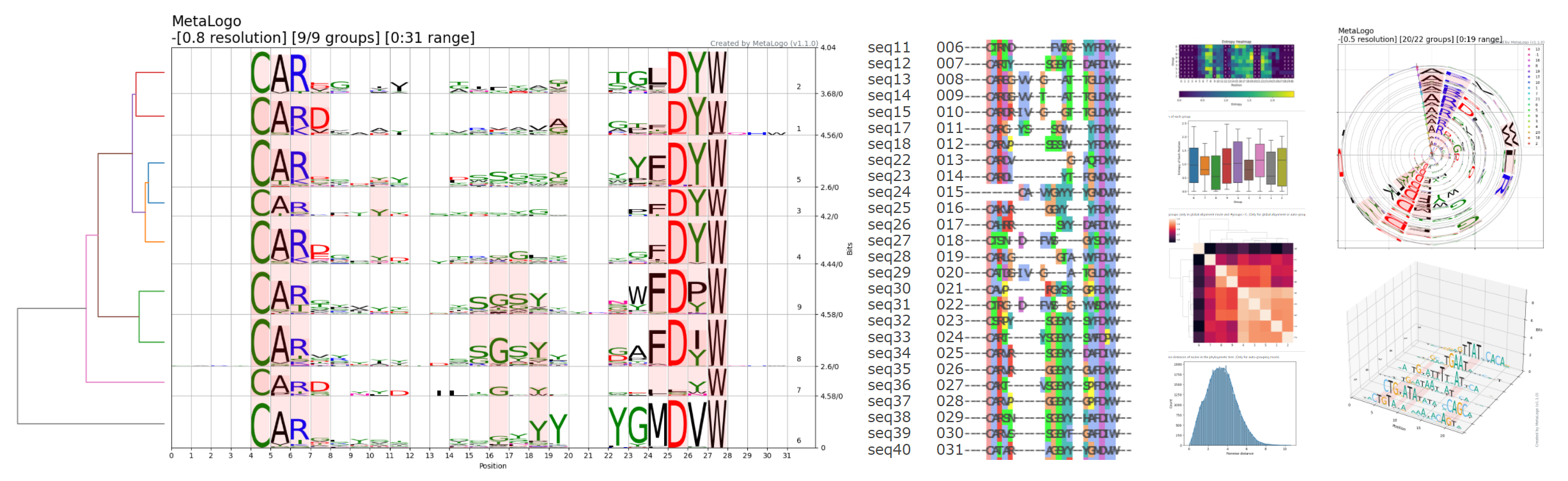
估计阅读时长: < 1 分钟MSA(多序列比对)在生物信息学中的核心目标是:通过把多条同源序列“对齐”,来突出它们之间的相似与差异,从而帮助我们:识别保守区/功能位点、推断进化关系(系统发生)、预测或解释蛋白质/核酸结构、发现共进化与功能模块,以及为后续分析(如模体搜索、结构建模、从头设计等)提供基础。基于多序列比对分析,我们可以通过这种算法,把一堆表面看上去“乱糟糟”的序列,整理成一个可以“逐位点比较”的框架。基于我们所得到的这个框架基础,我们可以进行下游的后续分析,例如: 识别哪些部分是“不能动”的(功能/结构核心); 推断它们是如何“进化而来”的(系统发生); 推测它们在空间中“长什么样”(结构预测与建模); 找出哪些部分“一起变化”(共进化与功能耦合); 并把这些信息封装成模型(HMM、profile)用于大规模搜索与注释。 Attachments MSA • 174 kB • 58 click 2026年1月8日
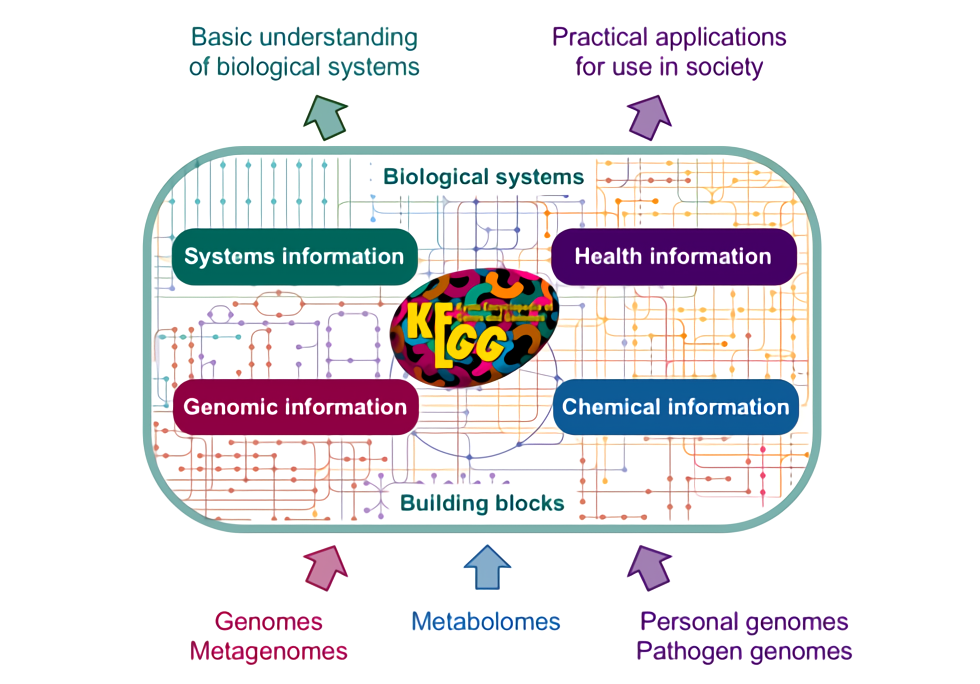
估计阅读时长: 16 分钟KEGG 里面目前并没有“现成的每个 KO 一条代表性序列 FASTA”这种官方序列数据库,假若我们需要基于KEGG数据库中的KO信息的注释,那我们一般会需要自己从 KEGG GENES 里面把每个 KO 对应的基因/蛋白序列抓出来,再按 KO 编号组织成 fasta 集合构建出对应的数据库。基于所建立好的KEGG基因序列数据库,我们就可以实现下面的一些基因注释工作: 在全基因组规模代谢网络重建工作中,进行我们的目标基因组中的代谢网络中的酶节点的直系同源推断,从而将我们的目标基因组中的基因映射到具体的KEGG代谢网络上的节点位置,从而重建出代谢网络模型(使用带有KO编号的蛋白序列做比对注释) 假若我们在进行宏基因组的基因丰度的计算,则可以基于所建立的KEGG基因序列数据库作为参考库,进行宏基因组测序数据中的KO基因丰度的计算(使用带有KO编号的基因序列做比对注释) […]

Wow that was strange. I just wrote an extremely long comment but after I clicked submit my comment didn't show…
Hi, appreciate the effort put into this. It's always good to see quality content. 🥳
WOOOOOW This was incredibly helpful and easy to understand. I've learned a lot. many thanks to your idea sharing.
[…] 在前面写了一篇文章来介绍我们可以如何通过KEGG的BHR评分来注释直系同源。在KEGG数据库的同源注释算法中,BHR的核心思想是“双向最佳命中”。它比简单的单向BLAST搜索(例如,只看你的基因A在数据库里的最佳匹配是基因B)更为严格和可靠。在基因注释中,这种方法可以有效减少因基因家族扩张、结构域保守等原因导致的假阳性注释,从而更准确地识别直系同源基因,而直系同源基因通常具有相同的功能。在今天重新翻看了下KAAS的帮助文档之后,发现KAAS系统中更新了下面的Assignment score计算公式: […]
不常看到, 没有多余矫饰的表达。敬意。